Post by: souro10 on December 18, 2012, 11:19:34 AM
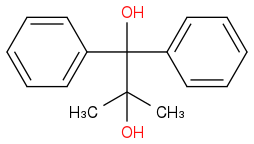 .
.We know that first the Phenylic Carbocation will be formed , followed by a methyl shift. However, my question is , if the reaction is carried at high temperature and for a longer time , would the thermodynamically more stable conjugated phenyl-ketone be formed ( which is formed through a less stable Methy carbocation intermediate , followed by Phenyl shift ) ?
I haven't found any textbooks talking about this. Thanks.
Post by: orgopete on December 18, 2012, 07:19:56 PM
Post by: camptzak on December 19, 2012, 01:25:55 AM
If the reaction moves to the ketone, couldnt the ketone again be protonated and undergo a methyl shift back to the cation stabilized by two phenyl rings?
Post by: souro10 on December 19, 2012, 11:39:39 AM
The driving force for Pinacol-Pinacolone rearrangement is actually the Octet configuration attainment.
My question is different. Orgopete , the dehydration can be the key-step , and one carbocation ( the resonance stabilized one with phenyl rings ) will be formed preferentially more than the other one. But why after longer reaction times , the reaction won't ( or will it? ) come under equilibrium / thermodynamic control? My understanding of kinetic v/s thermodynamic control is - when there are two pathways through two different intermediates having different energy of activation of their transition states in a particular reaction , and if it so happens that the activation energy of one intermediate is more than the other, but the corresponding final products are at a lower energy state (more stable) than the latter, then the latter at high temperatures begins to retrace its way back to the products and now it has sufficient energy to cross the activation energy barrier and reach the most stable product state. And this exactly happens in this case.
Hence, my question.
Post by: orgopete on December 19, 2012, 02:30:21 PM
Agreed. In most reactions, the kinetic and thermodynamic products are the same. In order for a different product to form after the kinetic product forms, there must be a reversible path to the thermodynamic product. Hence I argued about how the reaction should occur.
Since the question was whether more vigorous conditions could lead to a different product, I tried to follow two different reversible intermediates, the initial carbocations and the rearrangement intermediates. I would argue that mixtures are more common if the carbocations are nearly equal in stability, though that would not preclude faster migration being rate determining. In this instance, I would expect the doubly benzylic would be far more stable and thus limit formation of an alternate product on that account.
Now, I wasn't just trying to tell you the reaction couldn't give another product, but I wanted you to think methodically about the chemistry that may be useful in other problems. So, I addressed the question of the retro-pinacol rearrangement. One could form the same intermediate by beginning with 3,3-diphenylbutan-2-one and treat it with acid. Protonation would give the intermediate that formed after methyl migration. Certainly, I would expect this to be a rational step, for example in a ketalization of this ketone. If the methyl group were to migrate, it too could give the more stable intermediate formed by dehydration of the diol. I would argue two facets. One, protonation does not rearrange ketones by formation of alternate carbocations, I don't know of any examples right off. Secondly, if we were to write the resonance form of a protonated ketone as a carbocation, then we might argue the non-bonded electrons of oxygen are more stabilizing that two benzylic groups for an equilibrium carbocation in order for the rearrangement to reverse.
If this analysis is correct, then you may try to bias reaction conditions or you may search for examples of acid catalyzed rearrangements, for example during ketalization reactions. I would expect you may find it occurs with alpha hydroxy ketones, for example. Just saying'.
Post by: souro10 on December 19, 2012, 04:47:10 PM
However, it contradicts my understanding of Thermodynamic v/s kinetic reaction control.
When you say " in order for a different product to form after the kinetic product forms, there must be a reversible path to the thermodynamic product " => there must be a reversible path back to the reactants , and then at the raised temperature, sufficient activation energy is there for the thermodynamic fav. product to form. Fine. But when you say reversible path , you essentially mean a plausible chemical mechanism ( in context of organic chemistry ) must exist to revert to the reactants.
But many books, and many websites do not say anything about the chemical reverse pathway. What they say is, and what I had initially understood is - after the kinetic product is formed, "at some slightly higher temperature, the first reaction will become reversible while reaction leading to thermodynamic product still remains irreversible. So although P1 may form initially, over time it will revert to starting material and react to give the more stable P2, the thermodynamically favored product "
What seems reading this is , the high temperature causes a lot of increase in energy in the reaction mixture, and the reactants move back to the hill-top of activation energy , and back to the starting materials. They don't mention about the essential fact that a plausible reaction mechanism to revert to the starting material must be there.
Your take on this, Orgopete?
Post by: orgopete on December 19, 2012, 08:27:15 PM
Thanks, thanks a lot for your explanation.
However, it contradicts my understanding of Thermodynamic v/s kinetic reaction control.
When you say " in order for a different product to form after the kinetic product forms, there must be a reversible path to the thermodynamic product " => there must be a reversible path back to the reactants , and then at the raised temperature, sufficient activation energy is there for the thermodynamic fav. product to form. Fine. But when you say reversible path , you essentially mean a plausible chemical mechanism ( in context of organic chemistry ) must exist to revert to the reactants.
But many books, and many websites do not say anything about the chemical reverse pathway. What they say is, and what I had initially understood is - after the kinetic product is formed, "at some slightly higher temperature, the first reaction will become reversible while reaction leading to thermodynamic product still remains irreversible. So although P1 may form initially, over time it will revert to starting material and react to give the more stable P2, the thermodynamically favored product "
What seems reading this is, the high temperature causes a lot of increase in energy in the reaction mixture, and the reactants move back to the hill-top of activation energy , and back to the starting materials. They don't mention about the essential fact that a plausible reaction mechanism to revert to the starting material must be there.
A shortcoming in this discussion is an attempt to compare this reaction in which the product is both the thermodynamic and kinetic with another reaction that gives different products under kinetic and thermodynamic conditions. In the addition of HBr to butadiene, the kinetic product is 3-bromobut-1-ene and the thermodynamic product is 1-bromobut-2-ene. I don't understand where the contradiction exists. The quote given specifies the reaction is reversible. However, it is incorrect that a reaction that can give different kinetic and thermodynamic products must go back to starting materials. In the addition to butadiene, the reaction only goes back to the resonance stabilized carbocation. The same carbocation gives different products. I rationalize that bromide adds to the secondary carbon as it is attracted the greater positive charge of the CH3CH(+)CH=CH2 cation and gives the kinetic product. However, the C-Br bond is easily broken to reform the same intermediate. If bromide were to react at the CH2-carbon, it will give a product with a more stable double bond because it is more substituted and a more stable C-Br bond because its carbocation would be less stable than its allylic secondary counterpart.
Post by: curiouscat on December 20, 2012, 12:19:46 AM
In order for a different product to form after the kinetic product forms, there must be a reversible path to the thermodynamic product.
@orgopete:
I was a bit confused about your usage of the term "reversible path". On a micro-scale aren't all mechanistic pathways reversible? Or do you mean by this that a pathway where the reverse barrier is not extremely high?
Post by: orgopete on December 20, 2012, 09:58:54 AM
@orgopete:
I was a bit confused about your usage of the term "reversible path". On a micro-scale aren't all mechanistic pathways reversible? Or do you mean by this that a pathway where the reverse barrier is not extremely high?
Re: reversible
I had not planned a semantic argument in this problem. I had thought just as souro10 had quoted from his book, see text in red. If an intermediate reacts further, there must be a path for further reaction. If it reforms an intermediate it had passed through, then it must have been reversible, no?
I agree that ultimately this is about energy levels. However, I don't know how to express that in meaningful terms other than to indicate the reaction reverses. A low barrier results in a fast reaction. I large difference makes a reaction irreversible. In the poster's retro-pinacol, if the reaction is irreversible, is it because the activation energy of the product is much higher or because it is much lower in energy than the alternate product suggested by the poster. That was the rationale of the poster. I was taking the position that if I didn't know (as in I don't know), then how might I answer this problem. Certainly there are chemists that specialize in calculating energy levels in attempts to explain that which is found in the laboratory. I am not among them.
Post by: souro10 on December 21, 2012, 10:58:43 AM
Post by: orgopete on December 22, 2012, 03:23:20 PM
Post by: souro10 on December 22, 2012, 04:41:12 PM
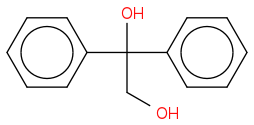
" The above example " (treatment of the above with H+ , fast ) " predicts kinetic versus thermodynamic product control. Under mild acid condition, the diol rearranges rapidly to an aldehyde via a 1,2-H shift to the initially formed diphenyl tertiary carbocation. More vigorous acid treatment of the diol or the aldehyde generates the more stable phenyl ketone ( a conjugate system of the phenyl and carbonyl groups ) "
Post by: souro10 on December 22, 2012, 06:12:35 PM
Under Pinacol rearrangement , check out Examples 1 and 2. Also check out the mechanisms they have shown by clicking. My argument is , by the same mechanism , shouldn't the more stable phenyl-ketone conjugate system be formed as the final product under drastic conditions - as i posted in my first post of this thread and this forum.
Post by: orgopete on December 23, 2012, 10:27:25 PM
It is also clear how the poster's question might arise as kinetic and thermodynamic products are shown for a pinacol rearrangement. Rearrangement of the aldehyde from a pinacol rearrangement can be driven to an alternate ketone. In fact, the poster's reactant is included in example #5 on the MSU website leading to two different products under different conditions.
At this point, I don't know what to say. The protonated aldehydes did rearrange. Will a protonated ketone also rearrange? How will the stabilities of the different intermediates affect the rates? Will the reaction of an aldehyde follow the same mechanism as that of a ketone? Will a protonated aldehyde be more attractive to accepting a neighboring group? If migration were to occur, does an aldehyde proceed by a loss of a proton or a hydride shift?
I was arguing from the very simple point of view that ketone protonation reactions are not uncommon. I was unaware of any notable rearrangements occurring. I think aldehydes react generally similar. I didn't think that even the dimethyl diphenyl diol presented a strong case for an alternate rearrangement. I can only speculate on whether a further migration of the methyl ketone can be made to occur upon protonation (assuming you would agree that is the reaction path). Since the question also asked whether more virorous conditions could lead to a different.product, then one might try the acetic anhydride modified pinacol as shown in the MSU reference. However, I don't know that different reaction conditions qualify as a description of a kinetic vs thermodynamic control of reaction products. A carboxylic acid plus alcohol can give an ester with acid catalysis. That is the thermodynamic product. An ester plus water will give a carboxylic acid and an alcohol with acid catalysis. That is also a thermodynamic product. It is the change of conditions that leads to the different products, and not one affected by the reaction rate, that is a fast forming intermediate that can be captured before formation of a more stable product.
Post by: souro10 on December 25, 2012, 06:19:12 AM
I agree that example 5 on MSU is not an example of kinetic v/s thermodynamic control, because what acetic anhydride basically does is - it attacks at the less hindered site, to produce the less stable initial dimethyl carbocation, which rearranges and stuff.
However, look at the first example. Or the one that i posted in my last reply. Here, the website, and some books claim that thermodynamic v/s kinetic product control exists. The examples listed on MSU are taken directly from the Solomons and Fryhle's O-Chem book. I posted the MSU link since it was inconvenient for me at that moment to scan and upload the pages. Now, the Solomons text book has the exact same examples- but the mechanism is not worked out - while MSU has given a mechanism, but I have serious doubts about its correctness.
An important thing to note is , Kinetic v/s thermodynamic control has been shown to occur only when there is hydrogen as a substituent on the beta carbon. None of the other cases, including the one I originally posted in my first post have been reported/stated anywhere to show KFP V/s TFP.
Now, this intrigued me big time. From your discussion, and also after reading many articles and chapters on Thermo v/s Kinetic control, I got it clear that for an alternative TFP to exist , there must be a plausible reaction mechanism from the TFP to the KFP, otherwise how will the initially-formed-faster-major-product get converted to the TFP?
Standing here, what I feel is , the mechanism shown on MSU is not correct. It contradicts my understanding of chemistry. I mean, why will a protonated carbonyl oxygen lead to a carbocation? I mean even if one applies Resonance, the resonance contribution by the carbocation will be way too small in contrast to the octet-stabilized protonated carbonyl. So a reverse reaction even if it occurs, will be exceptionally slow if that is really the mechanism. And I don't so why such a reaction should at all take place!
I think a possible and more logical explanation can be given with keto-enol tautomerism . In example 1, initially an aldehyde is formed. But it is an rapid equilibrium with its enol form. Now, normally the -OH of an enol form does not get protonated because it would lead to a highly unstable vinylic carbocation. However, in this case I will argue that protonation would still occur, but that would not lead to a carbocation, but a resonance stabilized Phenonium ion - Neighboring Group Participation by the Phenyl ring. Now, when we visualize the phenonium ion transition state, the bonding is unsymmetrical , and more amount of partial positive charge is present on the carbon atom which is attached to two phenyl rings.
The molecule(s) of water lost in course of the reaction can attack using their lone-pairs on oxygen at the delta-plus end, resulting in the expected phenyl ketone.
This is my explanation/ how I would like to justify the reaction. Once again, I think the mechanism on MSU for Example 1, is highly improbable.
Post by: orgopete on December 26, 2012, 12:51:58 AM
Although I hadn't looked at the MSU mechanisms, until disputed, I thought it fit the data and seemed plausible. Protonation of aldehydes and ketones are common intermediates in many mechanisms, for example ketalization or enolization reactions. How would you propose an acid catalyzed enol formation without protonation?
Post by: souro10 on December 26, 2012, 08:10:36 AM
However, my point is different. What I meant was, the carbonyl oxygen can get protonated. It can form resonance structures, and can lead to enolization/ketalization. However, none of those reactions, that I've heard of which occur via protonation of carbonyl oxygen, result in a 1,2 carbocation rearrangement. A carbocation rearrangement required sufficient , full positive charge - an actual carbocation to be formed first. Only then it can undergo rearrangement (if it has scope). I don't see how protonation , in this case would lead to carbocation formation.
Can you?
Post by: orgopete on December 26, 2012, 10:43:17 AM
Post by: souro10 on December 26, 2012, 07:19:00 PM
My question is, precisely , is - is it plausible to think/how is it justified that the protonation of a carbonyl oxygen can lead to sufficient positive charge on the carbonyl carbon for a carbocation 1,2 shift to take place, as depicted on MSU website? In my opinion it is not plausible because there would not be sufficient energy for the rearrangement to take place. Recall why rearrangements do not occur in SN2 reactions. Why? Because there is delta plus- not sufficient energy.
Can you post some other literature/ reference which depicts the mechanism shown on MSU website ( for example 1 ) , hopefully with more clarity and reasoning?
Post by: orgopete on January 01, 2013, 03:14:36 PM
@Orgopete - Sorry, I could not understand what you meant.
My question is, precisely , is - is it plausible to think/how is it justified that the protonation of a carbonyl oxygen can lead to sufficient positive charge on the carbonyl carbon for a carbocation 1,2 shift to take place, as depicted on MSU website? In my opinion it is not plausible because there would not be sufficient energy for the rearrangement to take place. Recall why rearrangements do not occur in SN2 reactions. Why? Because there is delta plus- not sufficient energy.
Can you post some other literature/ reference which depicts the mechanism shown on MSU website ( for example 1 ) , hopefully with more clarity and reasoning?
Re: rearrangement reaction in SN2 reactions
Remember, SN2 reactions are on the nucleophile initiated end of the substitution spectrum. In SN1 reactions, bond breaking occurs before bond formation. In SN2 reactions, it is the opposite, bond formation precedes bond cleavage. The bond formation increases electron density on the target carbon, it would be unusual for a rearrangement to occur. If a reaction were to be more SN1-like, then you may see this happen. Reactions can be thought to occur somewhere between the two extremes I have noted.
Re: ability of a carbonyl carbon to attract electrons, rearrangement or addition
My argument was simple, although it is unusual for this reaction to take place, it should be taken in context of all carbocations. Granted that non-bonded electrons of an attached oxygen will limit the attractiveness to that carbon, but this should also be taken in context of are there any attacks? Yes, ketalization and hydrolysis involve direct attack upon a carbonyl carbon. Even an enolization must be an attraction of the C-H electrons to the carbon.
Perhaps it is unusual for a carbon to migrate, so the instances are limited to groups that can provide greater stabilization that normal, such as the diphenyl case. I also noted that Reusch suggested that a protonated epoxide may be considered. I'd also concede that though Reusch did not give any compelling examples to argue this point.
This is my perspective upon this question and chemistry in general. Our models are not complete. It isn't possible to have partial charges, for example. Electrons are negative and protons positive and only involve full charges. If you compare the oxygen of hydroxide, water, and hydronium ion, the oxygen is +8 in all instances. What changes is the number of protons attached to the electrons. They have an effect upon the electrons and their availability, but no effect upon their charge. If you use this perspective, then you may look at reactions a little differently. It isn't whether a functional group should react in a certain way, it is how the assemblage of atoms may affect the electrons. For example, you may find some books write a hydroboration reaction as though it were a (symmetry forbidden) 2+2 reaction and HCl does not. Borane is a Lewis acid and is attracted to the electrons of an alkene. What I ponder is how this should affect a reaction, for example, boron holds its electrons much more weakly and its bond lengths are greater.
I have included a scheme and I suggest it should not be unusual to see attack upon a protonated carbonyl at the carbon atom. Enolization can be thought as a neighboring group reaction. The rearrangement may not be a general reaction (hence my error in assumption about it). I think the examples given are consistent with other chemistry. For example, protonation of an enol ether can be written with the same intermediate as the enolization step. If that is correct, then migration of a phenyl group may also occur similarly, especially if it were to lead to a "stabilized carbocation". (I too found it difficult to think of an carbocation alpha to a carbonyl group to be more stable.)
Granted, mechanisms are rationalizations. I agreed with Reusch's mechanism. It seemed plausible and consistent.
Post by: souro10 on January 01, 2013, 05:56:26 PM
I agree to this mechanism of yours, however there still remains a doubt. In the second rearrangement in your scheme, that is the one with three phenyl substituents, how does the third intermediate( from the left ) reach the final( or the fourth) product ? By loss of a proton? That is by enol formation? The mechanism for this particular rearrangement on the MSU website ( example 2 ) is slightly different, which I considered not plausible. For both Examples 1 and 2 , they are showing that a rearrangement or hydride shift is occurring instead of a proton loss which would lead to enol formation.
Secondly, I'd like to know how you'd account for the fact that the C-label is scrambled, using your mechanism? Would you go for an epoxide intermediate ? How would you accommodate an epoxide intermediate with this mechanism, for the triphenyl case?
And lastly, can we now conclude - in Pinacol-Pinacolone rearrangements, if the initially formed product under mild acid treatment is an aldehyde, it can rearrange itself into a more stable ketone ( often, a phenyl-ketone conjugated system ), although rearrangement of ketones into more stable ketones have not been reported. How can we account for the fact that ketones ( as in my original question ) do not usually rearrange into ore stable ketones? Is it because the hybridization changes from sp2 to sp3( and bond angle decreases from 120degrees to 109.5 degrees) in the migration-transition-state and hence for ketones steric factor kicks in? I think there could be further factors which does makes ketones rearranging into more stable ketones upon protonation uncommon. We must keep in mind the fact that when a Nucleophile attacks the carbonyl-carbon , it does so from an angle of approximately 107 degrees with the oxygen atom. Now during rearrangement of ketones due to the very nature of the rearrangement (anti-periplanar) , keeping such an angle of attack becomes more difficult due to sterric congestion by the already existing substituent.
I was going through " Acid catalyzed rearrangement of Aldehydes and ketones " from Jerry March's Advanced Organic Chemistry. From this book, and also the reference research papers mentioned at the bottom of the pages, it is obvious that the reaction involves an epoxide intermediate. And in the first step, it seems that there is simultaneous Neighboring Group Participation by oxygen and Phenyl !
Post by: orgopete on January 01, 2013, 10:59:38 PM
Re: hydride shift
I was expecting enol formation via deprotonation. Then I was looking at an abstract of a paper for the rearrangement of glyceraldehyde to pyruvaldehyde and dihydroxy acetone. Proton exchange with solvent did not occur to the methyl group, the label showed a hydride shift occurred. Although this could have been enol formation, the lack of incorporation of a label shows a hydride had migrated. Mechanistically, this should be the same reaction.
Re: ketone rearrangement reactions
I expect electronic effects to be controlling. As noted, diphenyl carbocations could form a reaction sink. None the less, migration may not occur to a neighboring tertiary carbon. The non-bonded electrons of oxygen would stabilize a carbocation. If the other substituent was a carbon rather than a hydrogen, it too would stabilze the carbocation and thus be less attractive for migration.
Re: stereo electronic effects
We already know a migration occurs to triphenylacetaldehyde, so any stereoelectronic barrier should be equal.
Returning to the original question, I think the alternate product could be formed. Whether it does or not may be another question. I know I have done reactions in which I had enough by-products to support many additional reactions. In order to answer that question, I'd look at the papers Reusch was citing. How good were the reactions, yield, by-products, etc. Low yields could account allow the reaction originally posted (in addition to others). If one wanted to find alternate products, obviously the pinacol is one low energy path, one can bias the conditions toward another reaction, as was the case here.
Sorry for the long-winded coming to an agreement with your original proposal. A case of too much ignorance and sloppiness.