This is the way I see it. Please excuse if I’m writing stuff you already know. I am trying to be as clear as possible, both for myself and for you.
Say you have a substance A that converts to B. Now if you mix A and B together at a certain temperature T, what happens?
A
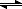
B
If you start with all A, you know you'll begin to form some B after some time passes. If you start with all B, you know you'll start to form some A. At equilibrium (infinite time), you know the relative concentrations (ratio) of A and B will be the same regardless of whether you start with all A and all B. This ratio is specified by the equilibrium constant, K. And K will be dependent on the temperature.
We introduce the concept of ΔG°, the Gibbs energy of formation. This is equal to the difference between the standard Gibbs energy of formation of B and the standard Gibbs energy of formation of A. In turn, the standard Gibbs energy of formation of each substance is the amount of free energy gained or lost from the formation of that substance at temperature T. These values are in turn expressed relative to the standard form of the substance. And they will be dependent on T because the amount of energy required to form a substance at a particular temperature is obviously related to T. Thermodynamic values are almost always expressed relative to other values.
Another way we can calculate ΔG° is from the equation
ΔG° = ΔH° – TΔS°
The change in the Gibbs energy of formation is temperature dependent, but most of the time it is expressed at 298.15 K because this is usually the value at which ΔH° and TΔS° are measured. (Although, ΔH° and TΔS° are usually taken as temperature independent quantities, so in practice it doesn’t matter, let’s be pedantic here and say it does.)
Now again, at temperature T if we start with concentrations of A and B at exactly equal to the ratio specified by the equilibrium constant at that temperature, then we are already at equilibrium at that temperature and we will have no net conversion of A to B or B to A. (We will see in a minute this is the case where Δ G = 0 because there is no net driving force in either direction.)
But let’s say we start with an A to B ratio (at temperature T) that is LARGER than the ratio specified by the equilibrium constant. In this case Q, the reaction quotient, is smaller than K (Q = [ B]/[A]). We know by intuition at this point that if this happens, we will get a spontaneous net conversion of some of the A into B, and this will continue until A and B are at the magic ratio specified by the equilibrium constant. The farther away the relative concentrations of A and B start from this equilibrium ratio, the greater this driving force for conversion will be. Also, if we start with an A to B ratio that is SMALLER than the ratio specified by the equilibrium constant, Q is larger than K and we will get a spontaneous net conversion of some of the B into A, and again this will continue until we reach the equilibrium ratio of [A] to [ B], specified by K, at temperature T. The equilibrium point will be the same regardless of the starting point!
Let’s introduce ΔG now. We’ve said that depending on the real ratio of A to B, with respect to the equilibrium ratio A to B, the reaction will tend to move one way or the other spontaneously. The direction of this movement, and the magnitude of the driving force, is embodied in ΔG . ΔG is dependent on the instantaneous state of the system at time t, where the concentrations of A and B are [A] and [ B], and the temperature is T. When the [A] and [ B] are far from the equilibrium value, the magnitude of ΔG will be large; when [A] and [ B] are at the equilibrium ratio, the magnitude of ΔG is zero.
We know that
ΔG = ΔG° + RT ln Q
As we’ve said, ΔG is a measure how spontaneous the consumption of A to B is (or B to A, depending on the sign of ΔG ) and is equal to zero at equilibrium. In a lot of textbooks on this subject they present a hypothetical plot of ΔG as a function of the mole fraction of one of the substances A or B (for a given T), which has a parabolic kind of shape. Where the minimum of this plot would be in relation to pure A or pure B is related to ΔG°. So ΔG° is a description of how how far to the left or right a reaction is at equilibrium. (As an example, if K = 1, then the concentration of A and B are equal at equilibrium, ΔG°is zero and the minimum of this hypothetical plot of ΔG vs. Q would be at a value of 1, because Q = K at equilibrium.
At equilibrium, then, ΔG = 0 and Q = K, so ΔG° = - RT ln K. In the case that K = 1, ΔG° = 0 because the equilibrium concentrations of A and B favor neither A or B (there are equal amounts of both). In this case ΔG for any given concentration of A and B just is = RT ln Q. What this essentially means is that if [ B] = 2[A], the magnitude of ΔG is the same as if [A] = 2[ B] (but opposite sign). This makes perfect sense because the driving force should be the same, but in opposite directions, along the chemical potential surface if the concentrations of A and B are switched - the equilibrium always will drive to a 50:50 mixture.
The final thing to consider is the effect of temperature. Temperature changes things only by shifting the equilibrium. The ΔG° value will also change if it is determined at a different temperature. In principle, you would need to determine a new ΔG° by either looking of Gibbs energies of formation at the different temperatures or by looking up enthalpies and entropies of formation at the different temperatures. The Gibbs energies of formation specify the free energy change for forming a given substance relative to the standard state of that substance, and this will depend on the temperature. In practice, however, it is almost always assumed that ΔH° and ΔS° are temperature independent, so what you have to do most of the time to determine ΔG° at a new temperature is to just use the same values of ΔH° and ΔS° and use the new temperature in the familiar equation above. You can then use this ΔG° value to determine a new equilibrium constant (- RT ln K) at this temperature and all the ΔG values for any given reaction quotient.
However the typical way to measure equilibrium constants at a new temperature is to just use the van't Hoff equation. (This is what your manipulation in the previous post was driving at, but you were trying to compare two temperature values with only one equilibrium constant.) When you do this you will see all that you really need is ΔH°, which as usual we've assumed to be temperature independent.
ln (K
2/K
1) = -(ΔH°/R)(T
2-1 - T
1-1)
Using this you can calculate K at any temperature as long as ΔH° is known at K is known at one temperature (typically at 25 °C, which is the easiest to calculate from tabulated data). Of course, the accuracy of this approach dependence on the variability of the enthalpy and entropy change wrt to temperature. Otherwise you need to do it the long way.
In the present problem, equilibrium works the same way, but you can't use concentrations for the equilibrium constants. You'll have to find some other way to express activity. As I said, I'm not sure of the appropriate way to do that.
Hopefully that is helpful and less confusing that previous discussion? Given my track record lately I'm sure there will be some kind of error in there somewhere that I'll have to later apologize for, but here's keeping my fingers crossed.

 ChemBuddy |
ChemBuddy |  Topic: Gibbs' Free energy Olympiad Question (Read 19051 times)
Topic: Gibbs' Free energy Olympiad Question (Read 19051 times)