Technicalhuman -
Practically speaking, ΔG° is basically a reference value which specifies what the relative concentrations of products and reactants are at equilibrium for a particular temperature. As such, ΔG° can be related to the equilibrium constant by the equation ΔG° = -RT ln K. Recall that the equilibrium constant is essentially determined by the relative thermodynamic activities/potentials of the reactants and products, which is why it can be related to the Gibbs energy (also an expression of chemical potential). As a reference value for potential energy, ΔG° can also be determined by heats of formation and entropies of formation (themselves determined relative to reference values), which is what your problem wants you to do. In other words, you can predict the value of the equilibrium constant for a process just by knowing the amount of heat energy released/absorbed through breaking/forming of bonds as well as the change in entropy, which itself is sort of a measure of the difference in the number of ways the available heat energy can be distributed.
So, to sum up here, you calculate ΔG° from ΔG° = ΔH° - TΔS°, and you calculate ΔH° and ΔS° in the usual way, using tabulated values for heats of formation and entropies of formation using a Hess law formalism.
Once you have ΔG°, this serves as a reference point for any other condition that the system may find itself in. Let's say for a simple reaction A
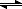
B, the equilibrium constant (at temperature 298 K) is 10. This means that at equilibrium, at 298 K, the concentration of B is ten times what the concentration of A is (assuming we can approximate the activites as concentrations, which is generally the case in solution). Using the equation ΔG° = -RT ln K, we can also determine that ΔG° for this reaction at this temperature is -5708 J/mol. This tells us that the conversion of A to B at room temperature will tend to be exergonic, that is, A has a higher chemical potential than B. Most of the time we do things in the opposite direction, though: if you start with ΔG°, calculated from heats and entropies of formation, you can learn what the relative values of A and B are once equilibrium is reached, at 298 K. You can usually easily get an estimate of ΔG° from tabulated thermodynamic reference values, and hence estimate what the equilibrium constant is likely to be.
What information we really want to know as chemists, however, is this: if I put a certain amount of A and a certain amount of B in a reaction pot, what is likely to happen? This is where ΔG comes in. From ΔG°, we can determine what the relative concentrations are going to be for A and B at equilibrium, and we already know, from above, that [B.] will be ten times that of [A]. We can guess from intuition that if we put a higher relative concentration of B in the pot than 10:1, we're going to generate more A (reaction will go backward), because we know the system is always going to approach equilibrium and form a 10:1 ratio, and the only way to do that is to reduce the amount of B and increase the amount of A. If we put a lower relative concentration of B in than 10:1, we are going to form more B - reaction will go forward. We express the real relative concentrations of B to A as the reaction quotient, Q. And from Q, using the reference value of ΔG°, we can determine a ΔG, which is the potential energy of the system when the reactants and products have any particular relative activities. ΔG quantifies the expected reaction direction.
So if we start out with a ratio of B to A of 5, Q = 5 and thus (using the ΔG° value above and the equation ΔG = ΔG° + RT ln Q) ΔG = -1718.3 J/mol. A negative ΔG means the specified process (conversion of A to B under the conditions specified) is exergonic and will happen spontaneously. Which makes sense: when the ratio of B to A is 5, this means we have less B (relative to A) than we know we should have at equilibrium. To go toward equilibrium, we need to form more B, so there should be a spontaneous formation of A to B. Hence ΔG we expect to be negative for A
B.
On the other hand, if we start with a ratio of B to A of 20, Q = 20 and ΔG = 1717.49 J/mol. A positive ΔG means the specified process (conversion of A to B under the conditions specified) is endergonic and will not happen spontaneously. This also makes sense: when the ratio of B to A is 20, this means we have MORE B (relative to A) than we know we should have at equilibrium. To go toward equilibrium, then, we need to form more A, so there should not be spontaneous formation of A to B (in fact, there should be spontaneous formation of B to A). Hence ΔG we expect to be positive for A
B.
Of course, if we start with a ratio of B to A of 10, Q = 10 and ΔG = 0. This shouldn't be surprising because we are already at equilibrium! There's no driving force to convert A to B or B to A.
So you see, ΔG gives us a real measure of the thermodynamic driving force (difference in potantial energy between reactants and products) for any particular set of starting conditions. ΔG° is a reference value that basically specifies what the equilibrium point is. By calculating ΔG, it will be possible to predict how the system will behave - is more A likely to be formed or is more B likely to be formed, relative to the starting point, once equilibrium is reached? Note also that the system is dynamic: Q will tend to change until it reaches the equilibrium constant K, at which point ΔG is zero and no more gross change will be observed... unless the system is disturbed (temperature change, more A added, etc). Note also that ΔG doesn't say anything specific about the
rate at which equilibrium will be reached, because kinetics is not determined exclusively by thermodynamic driving force.
I hope this clears up the difference between ΔG and ΔG°. ΔG is equal to zero when the system is at equilibrium, but ΔG° is a reference value that doesn't change for a specific set of conditions (temperature, e.g.).
 ChemBuddy |
ChemBuddy |  Topic: Gibbs Free Energy and G=Go+RTlnK derivation (Read 57470 times)
Topic: Gibbs Free Energy and G=Go+RTlnK derivation (Read 57470 times)