Hello everyone,
I have a question regarding molecule similarity computation. I'm more from computation than chemistry, so it is a fairly new topic for me, and I'm actually working with a (quantum computing) algorithm for molecule similarity computation.
So here is my question, given these molecules:
- niacin
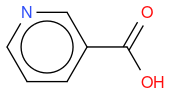
, hereafter "reference molecule"
- 4-CARBOXYPIPERIDINE
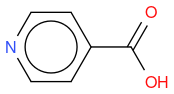
, hereafter "molecule 1"
- nicotinamide
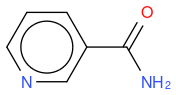
, hereafter "molecule 2"
- P modified nicotinamide
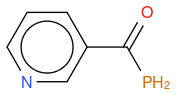
, hereafter "molecule 3"
If I compute the Tanimoto similarity between reference and molecule 1, I have 0.419.
If I compute the Tanimoto similarity between reference and molecule 2, I have 0.633.
What I observe is that Tanimoto similarity considers that molecule 2 is more similar to reference molecule than molecule 1, but if we look at molecule illustrations, we notice that molecule 1 differs from reference by one N atom moved by one position, whereas molecule 2 differs from reference by one molecule which is not the same.
So, in an algorithmic point of view, it makes sense that molecule 1 has two molecule differences (one N replaced by C, and one C replaced by N) whilst molecule 2 has only one molecule difference (OH replaced by NH2) so the similarity is lower for molecule 1.
But, in a chemical point of view, does this also make sense ? I mean, why just moving one N atom is less similar than changing one atom by an other ? In other word, is the chemical function of molecule 2 more similar than molecule 1 to reference molecule ?
An other observation, if I compute the Tanimoto similarity between reference and molecule 3, I have 0.633 (like for molecule 2), so Tanimoto distance does not take in account the fact that one atom differs between molecule 2 and 3, whilst my "non-chemical-specialist" mind would guess than one is more similare than the other as they are not equivalent ?
Finally, is there a "chemical" process (by chemical, I mean not algorithmic) to compare molecules in order to have "chemical function" similarity I can refer to ?
Thank you for your help, I hope my questions are well formulated.
 ChemBuddy |
ChemBuddy |  Topic: Molecule Similarity (Read 2598 times)
Topic: Molecule Similarity (Read 2598 times)